
Teilen auf
Twitter Facebook LinkedIn WhatsApp„Iss weniger Kohlenhydrate” ist heute so etwas wie die universelle Empfehlung für übergewichtige und stoffwechselkranke Menschen. Dahinter steckt die Idee, dass Zucker unseren Insulinspiegel in die Höhe treibt und dieser Insulinspiegel unsere Zellen auf Dauer insulinresistent macht. Wie genau der Insulinspiegel das anstellt, weiß man nicht. Aber es wird schon stimmen, schließlich machen Low Carb und Ketose die Menschen schlanker, fitter und gesünder.
Stimmt das wirklich? Dieser Frage geht der heutige Artikel auf den Grund. Achtung: Es folgen biochemische Details, die den Unterschied zwischen Fett- und Zuckerstoffwechsel herausstellen. Wenn Du nicht mit der Zellatmung vertraut bist, empfehle ich Dir als Einstieg Besser leben mit mehr Zellenergie.
Energiespeicher und -verbraucher
Auch wenn es uns manchmal nicht so vorkommt… Unser Stoffwechsel bildet ein intelligentes und fein austariertes Gleichgewicht aus anabolen und katabolen biochemischen Prozessen. Regisseur in diesem komplexen Spiel ist der Hypothalamus, der alle vegetativen Funktionen steuert. Für die Erläuterungen in diesem Artikel brauchen wir ihn allerdings nicht. Wichtiger ist das Verständnis von Fettgewebe, Muskulatur, Leber und Schilddrüse. Sie fungieren als Speicher, Verbraucher, Regler und Gaspedal im Energiestoffwechsel.
Fettgewebe: Speicher
Die Zellmasse des Fettgewebes besteht zu etwa 95 % aus Triglyceriden. Triglyceride sind drei Fettsäuren (Tri = drei), die an Glycerin gebunden (verestert) sind. Sie dienen als Energiespeicher. Bei Nahrungskarenz werden Triglyceride gespalten und ins Blut abgegeben. Die freigesetzten Fettsäuren werden von anderen Geweben aufgenommen und unter Energiegewinn abgebaut.
Bei einem Überangebot an Nahrung werden freie Fettsäuren und der Einfachzuker Glukose aus dem Blut aufgenommen und in Triglyceride umgewandelt. Fettsäuren können die Zellwand hormonunabhängig passieren. Die Aufnahme von Glukose in die Fettzellen erfolgt insulinabhängig (Bild unten).

Durch den Wechsel zwischen Lipogenese und Lipolyse nimmt das Fettgewebe aktiv am Stoffwechsel teil und macht uns unabhängig von kontinuierlicher Nahrungszufuhr. Das Fettgewebe eines normalgewichtigen Erwachsenen enthält etwa acht Kilogramm Triglyceride und deckt den Energiebedarf für 30 bis 40 Tage [1*].
Muskulatur: Verbraucher
Die Skelettmuskeln und der Herzmuskel sind neben dem Gehirn die größten Energieverbraucher im Körper. Schon mäßige körperliche Arbeit wie leichtes Joggen mit etwa 65 % der maximal möglichen Leistung lassen den Energieverbrauch um den Faktor 6 bis 8 ansteigen [1*]. Abhängig von der Stoffwechsellage verbrennen Muskeln sowohl Glukose als auch Fett, wodurch sie die Nährstoff-Homöostase des gesamten Körpers beeinflussen.
Um Glukose aus dem Blut aufzunehmen, benötigen Muskelzellen (wie Fettzellen) das Hormon Insulin. Bei längerer Aktivität oder starker Belastung verwerten Muskeln ihre eigenen Energiespeicher. Diese bestehen zu etwa gleichen Teilen aus Glykogen und leicht verfügbaren Muskel-Triglyceriden [2].
Leber: Regler
Die Leber ist das zentrale Organ des Zuckerstoffwechsels. Hier erfolgen der Ab- und Umbau von Kohlenhydraten und die Speicherung von Glukose als Glykogen. Etwa ein Drittel des im Körper gespeicherten Glykogens befindet sich in der Leber, die anderen zwei Drittel in der Muskulatur. Im Hungerzustand gibt die Leber Glukose ab, um die Blutzuckerkonzentration aufrecht zu erhalten. Bei Bedarf synthetisiert sie Triglyceride aus Glukose und Ketonkörper aus Fettsäuren. Diese werden ins Blut abgegeben und auf die verschiedenen Gewebe verteilt.
Schilddrüse: Gaspedal
Die Schilddrüsenhormone Thyroxin (T4) und das Trijodthyronin (T3) steigern den Sauerstoffverbrauch in nahezu allen Körperzellen. Weil T3 etwa viermal so wirksam ist wie T4, steigt die Stoffwechselrate proportional mit dem Spiegel an freiem T3 (fT3) im Blut. Mit dem gesteigerten Energieumsatz geht auch eine gesteigerte Thermogenese einher. Der Gegenspieler des T3 ist das Reverse-T3. Es kann bei Stoffwechselstörungen, Entzündungen und Krankheiten ansteigen [3].
Der Randle-Zyklus
Als Brennstoff für die Muskelzellen kommen sowohl Glukose als auch Fettsäuren infrage. Der Randle-Zyklus, auch bekannt als Glukose-Fettsäure-Zyklus, beschreibt die reziproke Beziehung zwischen Zucker- und Fettstoffwechsel:
Ein Überangebot an Fettsäuren blockiert die Aufnahme und Oxidation von Glukose in den Muskelzellen, im Gegenzug kann ein hoher Blutzuckerspiegel die Fettverbrennung verhindern.
Der Regulationsmechanismus wurde erstmals 1963 durch Philip Randle in “The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus” beschrieben [4].
Metabolisch gesunde Menschen können zwischen Fett- und Zuckerstoffwechsel umschalten. Je nach Substratangebot nutzen sie entweder Fette oder Kohlenhydrate als primäre Energiequelle. Der Schalter für diesen Switch ist das Insulin. Mit zunehmender Insulinresistenz verliert ein Mensch die Fähigkeit, Glukose zu verbrennen. Gleichzeitig kann er im nüchternen Zustand auch Fette schlechter verwerten [5].
Wie Fett den Zuckerstoffwechsel hemmt
Für die Glukoseaufnahme haben Fett- und Muskelzellen ein eigenes Transportprotein, den Glukose-Transporter-4 (GLUT-4). Unter Insulin-Einfluss wandert GLUT-4 aus dem Zellinneren in die Außenmembran. Glukose kann in die Zelle eindringen, wo sie zu Pyruvat und – unter aeroben Bedingungen – zu Acetyl-CoA abgebaut wird. Dieser Stoffwechselweg heißt aerobe Glykolyse. Das Endprodukt Acetyl-CoA wird im Citrat-Zyklus zu Citrat umgewandelt.

Bei der Fettsäureoxidation entsteht ebenfalls Acetyl-CoA. Ein Überangebot an Fettsäuren führt zu einer hohen Konzentration von Acetyl-CoA und Citrat in den Mitochondrien. Beide Substrate setzen eine Kaskade in Gang, die den Glukoseabbau an mehreren Stellen blockiert (Bild oben):
- Acetyl-CoA hemmt das Enzym Pyruvat-Dehydrogenase (PDH), das die Umwandlung von Pyruvat in Acetyl-CoA katalysiert.
- Citrat hemmt das Enzym Phosphofructo-Kinase (PFK), wodurch Glukose-6-Phosphat im Zytosol akkumuliert.
- Glukose-6-Phosphat in hohen Konzentration hemmt das Enyzm Hexokinase, so dass der Glukoseabbau zum Erliegen kommt.
- Zuletzt wird auch die GLUT4-Translokation in die Zellmembran gehemmt, möglicherweise durch Citrat oder Suppression des GLUT-4-Gens [6,7,8].
Weil die PDH am stärksten gehemmt wird (1), fällt zunächst weiter Pyruvat an. Dieses wird anaerob zu Laktat abgebaut, wodurch sich dass Zellmilieu zu sauren pH-Werten verschiebt. Erst wenn der Glukose-Einstrom blockiert ist (4), nimmt die Zelle trotz erhöhtem Insulin-Spiegel keine Glukose mehr auf.
Das Fatty Acid Syndrome
Je weniger Glukose in den Zellen ankommt, desto mehr Fettsäuren werden freigesetzt. Der Blutzucker kann stark ansteigen, weil die Glukoseproduktion in der Leber stimuliert wird. Randle bezeichnete diese Form der Insulinresistenz, die stark an Diabetes Mellitus Typ 2 erinnert, als “Fatty Acid Syndrome” [6,9].
Ein Biomarker für Insulinresistenz ist die Größe der intramuskulären Fettdepots. Mit Triglyceriden überfrachtete Muskelzellen können nicht mehr zwischen Fett- und Zuckerstoffwechsel umschalten. Der Grund sind Lipid-Metabolite wie langkettiges Acetyl-CoA, 3-OH-Butyrylcarnitin und Ceramide. Sie stören die Signalübertragung am Insulin-Rezeptor und senken die Insulinsensitivität [2,5,10].
Das Fatty Acid Syndrome – also die exzessive Oxidation von Fettsäuren – liefert eine plausible Erklärung für die Enstehung von Typ-2-Diabetes. Dass die Stoffwechselkrankheit durch Kohlenhydrate verursacht wird, ist wissenschaftlich nicht bewiesen. In einer koreanischen Studie stieg das Risiko für Diabetes, wenn Kohlenhydrate durch Fette ersetzt wurden [11,12].
Wie Zucker den Fettstoffwechsel hemmt
Ein hoher Blutzucker- und niedriger Triglyceridspiegel hemmt die Fettsäureoxidation und stimuliert die Lipogenese. Maßgeblich für diese Effekte sind die zellulären Wirkungen des Insulins (Bild unten):
- Insulin stimuliert den aeroben Abbau von Glukose zu Acetyl-CoA.
- Aus Acetyl-CoA entsteht Citrat, das ins Zytosol transportiert wird. Dort reagiert es in zwei Schritten zu Malonyl-CoA, dem Substrat der Fettsäure-Biosynthese. Insulin aktiviert das für die Malonyl-CoA-Produktion verantwortliche Enzym Acetyl-CoA-Carboxylase (ACC).
- Malonyl-CoA hemmt das Enzym Carnitin-Palmityltransferase 1 (CPT-1), das den Transport von Fettsäuren in das Mitochondrium katalysiert.
- Insulin stimuliert den GLUT-4-Transporter. Hierdurch steigt der Glukoseeinstrom in Herz- und Muskelzellen.

Die Hemmung der CPT-1 gewährleistet, dass bei der Lipogenese die Fettsäureoxidaton abgeschaltet ist. Die neu gebildeten Fettsäuren werden zu Triglyceriden verestert. Je höher der Anteil an Kohlenhydraten in der Nahrung ist, desto mehr Glukose wird in Fettsäuren umgewandelt.
Leberverfettung
Eine kohlenhydratlastige Ernährung fördert die Expression von ACC und anderen lipogenen Enzymen in der Leber, wodurch es zu einer verstärkten Neubildung von Triglyceriden kommt [6]. Wenn die Lipogenese langfristig die Mechanismen zur Entfernung der Fettsäuren übersteigt, akkumulieren die Triglyderide in den Leberzellen. Im Extremfall bildet sich eine nicht-alkoholische Fettleber.
Zucker- vs. Fettstoffwechsel
Wie wir gesehen haben, kann der Körper das für den Citrat-Zyklus benötigte Acetyl-CoA sowohl aus Glukose als auch aus Fettsäuren herstellen. Acetyl-CoA wird im Citrat-Zyklus zu Wasser und Kohlendioxid oxidiert, wobei NAD+ und FAD reduziert und anschließend in der Atmungskette reoxidiert werden. Beide Stoffwechselwege verlaufen damit weitgehend identisch (Bild unten).

Sie sind allerdings nicht gleichwertig! Die entscheidenden Unterschiede ergeben sich aus der Umwandlung von Glukose bzw. Fettsäuren in Acetyl-CoA.
Die Glukose-Oxidation produziert mehr Kohlendioxid
Bei der Oxidation von Kohlenhydraten entsteht 50 % mehr CO2 als bei der Oxidation von Fett. Grund ist der Respiratorische Quotient (RQ) beider Substrate. Dieser beschreibt das Verhältnis zwischen dem gebildetem bzw. abgeatmetem CO2 und dem vom Körper aufgenommenen O2 [13]:
Respiratorischer Quotient = VCO2 ÷ VO2
Der RQ für Kohlenhydrate ist größer als der für Fette, nämlich:
RQKH = 1; RQFett = 0,7
Wer die zugrundeliegende Berechnung nachvollziehen möchte, dem empfehle ich dieses 10-minütige Video von Jörn Loviscach.
Kohlendioxid erfüllt im Organismus wichtige physiologische Funktionen:
- Ein hoher CO2-Partialdruck sorgt für eine gesteigerte O2-Zufuhr in die Zelle (Bohr-Effekt). Die Zelle kann mehr ATP produzieren und die Stoffwechselrate steigt [14]. Der zusätzliche Sauerstoff spielt eine wichtige Rolle für die Energieversorgung des Gehirns [15].
- CO2 ist ein starker Vasodilatator, der die Gefäße weitstellt und die Sauerstoffzufuhr des Gewebes weiter erhöht [16].
- CO2 schützt die Zellen vor oxidativem Stress [17].
Fazit: Kohlendioxid verbessert die mitochondriale Atmung.
Die Fettsäure-Oxidation produziert mehr FADH2 und weniger NADH
NADH und FADH2 sind die wichtigsten Elektronenüberträger der mitochondrialen Atmungskette. Sie werden zu NAD+ und FAD oxidiert und ihre Elektronen für die ATP-Synthese genutzt. Beim aeroben Abbau von Glukose entstehen zwei Moleküle Pyruvat. Beide werden zu Acetyl-CoA abgebaut. Pro Molekül Acetyl-CoA werden zwei NADH produziert. Hinzu kommen 3 NADH und 1 FADH2 aus dem Citrat-Zyklus (Bild unten).
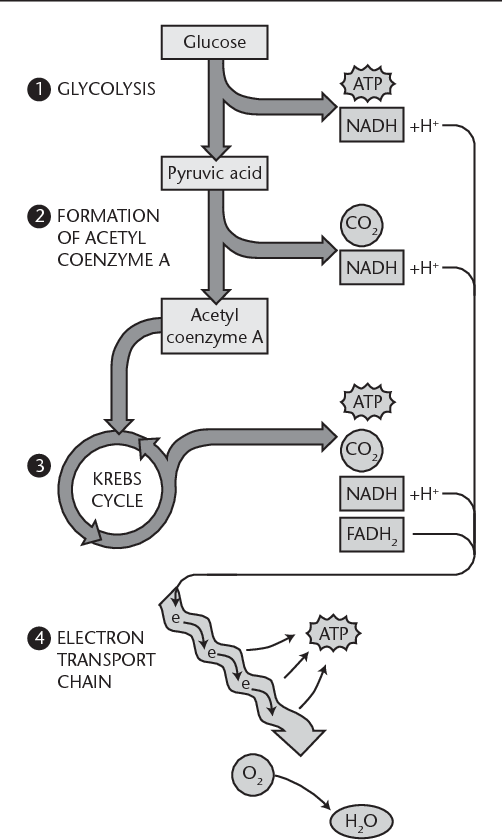
Bei der Fettsäure-Oxidation (β-Oxidation) werden Acetyl-CoA-Moleküle von der Fettsäure “abgeschnitten”, bis die Fettsäure vollständig zu Acetyl-CoA aufgespalten ist. Im Durchschnitt entstehen für jedes Acetyl-CoA ein NADH und ein FADH2 (Bild unten).
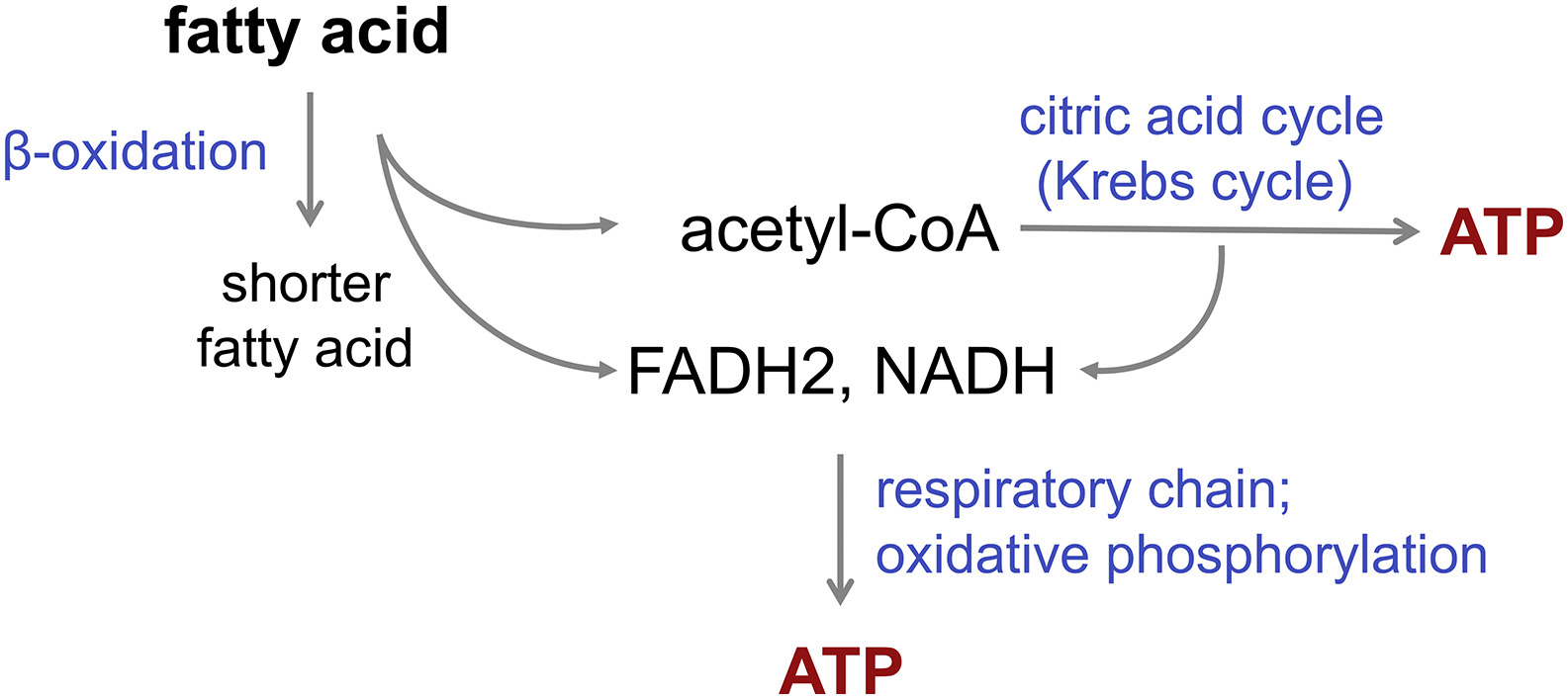
Beispielsweise werden bei der β-Oxidation von Palmitinsäure 15 FADH2 und 31 NADH produziert, was einem FADH2/NADH-Verhältnis von
FADH2 / NADH Palmitinsäure ≈ 0,5
entspricht. Beim vollständigen Abbau eines Glukosemoleküls werden zwei FADH2 und 10 NADH gebildet, das entspricht einem FADH2/NADH-Verhältnis von [20]:
FADH2 / NADH Glukose = 0,2
Das Verhältnis von FADH2 und NADH beeinflusst die mitochondriale Atmung. NADH spendet seine Elektronen im Komplex I der Atmungskette, während FADH2 seine Elektronen im Komplex II abgibt. Beide Komplexe konkurrieren um denselben Elektronenakzeptor: Ubichinon (Q10).
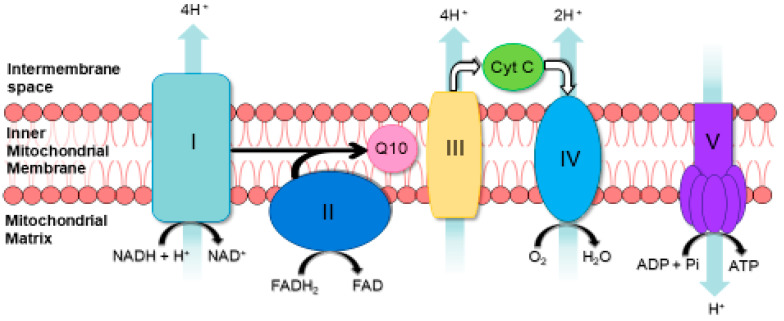
Ein hoher Quotient FADH2 / NADH setzt die nachfolgenden Reaktionen in Gang:
- FADH2 reduziert den Pool an verfügbarem Q10 am Komplex II, wodurch es zu einer Hemmung und Anhäufung von Elektronen am Komplex I kommt.
- Die Hemmung von Komplex I führt zur vermehrten Bildung von Superoxid, einem aggressiven Sauerstoffradikal und potenten Zellgift [22].
- Es kommt zu einem Anstieg des Quotienten NADH / NAD+. Dieser ist ein wichtiger Biomarker für eine gestörte Respiration [22].
Fazit: Zuviel FADH2 hemmt die mitochondriale Atmung.
CO2 und NADH sind der Grund, warum unsere Zellen Kohlenhydrate wesentlich effizienter verbrennen und dabei mehr Energie erzeugen als bei der Oxidation von Fettsäuren. Daraus folgt: Glukose ist das bevorzugte Substrat. Fette sind der Reservetreibstoff für Zeiten, in denen keine Kohlenhydrate verfügbar sind.
Fettstoffwechsel-Adaption
Im Fettstoffwechsel müssen unsere Zellen mit weniger ATP auskommen. Weil Energiemangel Stress verursacht, schüttet der Körper die Stresshormone Cortisol und Adrenalin aus. Sie halten den Blutzuckerspiegel durch Eiweißabbau aufrecht. Gleichzeitig senkt der Körper die Energieproduktion und den Energieverbrauch, indem er die Produktion des aktiven Schilddrüsenhormons T3 drosselt [23,24,25]:
Energiemangel ⟶ Stress ⟶ T3 fällt und rT3 steigt.
Die Folge: Der Stoffwechsel wird langsamer. Die gesunkene Stoffwechselrate bewirkt, dass wir nach einer längeren Fastenperiode oder Low/No-Carb-Diät umso schneller zunehmen, sobald wir wieder Kohlenhydate konsumieren. Der Körper speichert die zusätzliche Energie als Fettdepot (Jojo-Effekt).
Ketone für die Nervenzellen
Die meisten Körpergewebe können mit Fett als Brennstoff überleben. Nicht so das Gehirn – Unser energieintensivstes Organ benötigt Glukose und Ketone als Brennstoff. Ketonkörper werden bei längerem Hungern bzw. unter einer fettreichen und kohlenhydratarmen Ernährung (Ketogene Diät) gebildet [26].
Zuerst werden allerdings die Glykogenspeicher aufgebraucht. Wenn diese leer sind, stellt die Leber Glukose aus Aminosäuren her (Glukoneogenese). Dabei wird sowohl Nahrungsprotein als auch körpereigenes Protein verbraucht [27,28]. Weil die Glukoneogenese Oxalacetat benötigt, entsteht ein relativer Mangel an Oxalacetat, so dass Acetyl-CoA nicht mehr in den Citrat-Zyklus eintreten kann.
Weil die Fettsäureoxidation auf Hochtouren läuft, akkumuliert Acetyl-CoA in den Leberzellen. Die Leber wandelt es in den Mitochondrien in die wasserlöslichen Ketonkörper Acetoacetat und 3-Hydroxybutyrat um:
2 Acetyl-Coa ⟶ … ⟶ Acetoacetat ⇌ 3-Hydroxybutyrat (+ Aceton)
Anschließend gibt sie beide ins Blut ab, wo sie extrahepatischen Geweben als Energieträger dienen (Bild unten).
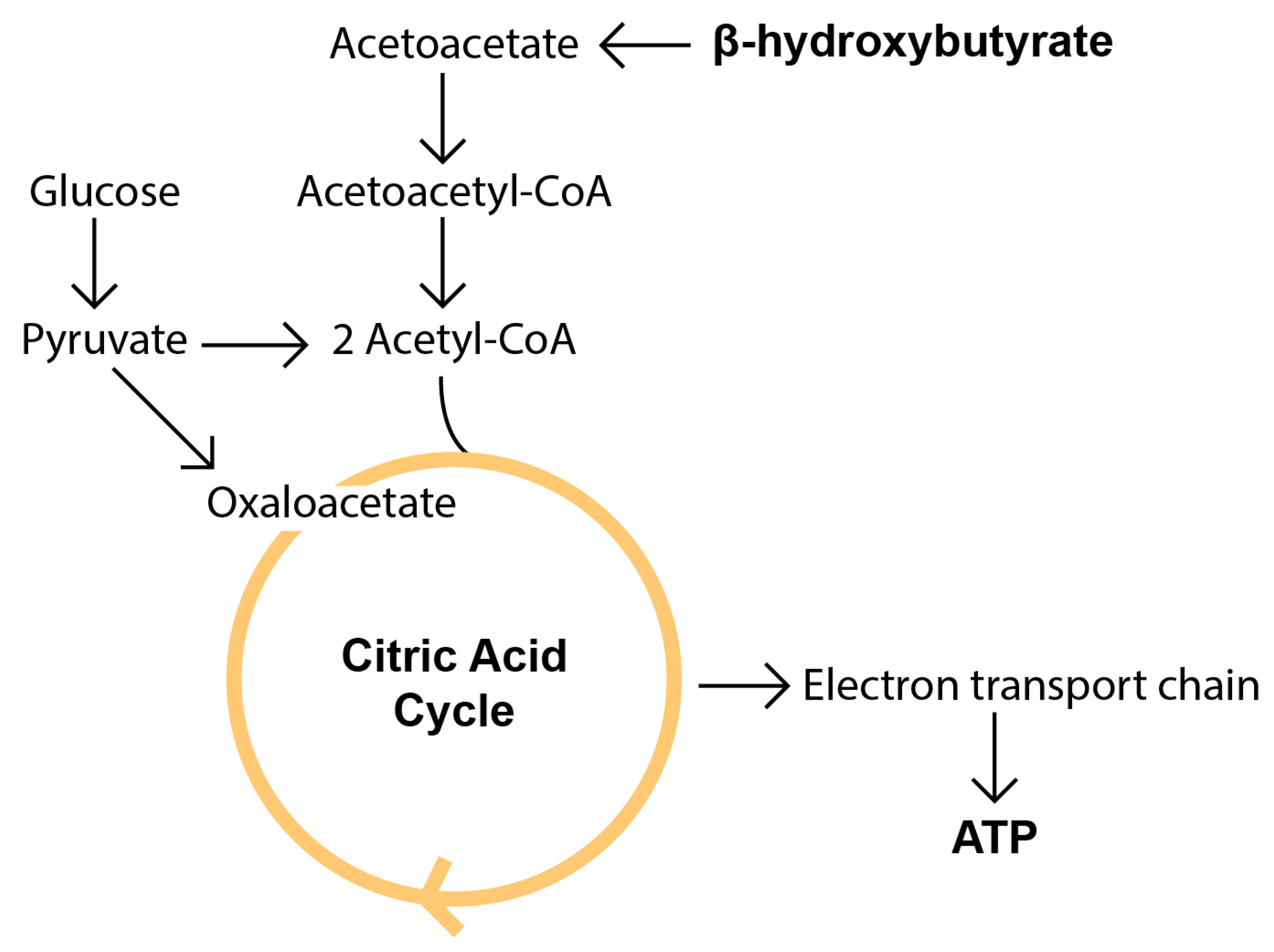
Das Gehirn kann ca. 75 % seines Brennstoffbedarfs aus Ketonkörpern decken, der Rest muss mit Glukose versorgt werden. Die Hoden, das Nierenmark und die Erythrozyten funktionieren ausschließlich mit Glukose. Daher wird die Glukoneogenese während einer ketogenen Diät aufrecht erhalten [30].
Diabetes Mellitus Typ III
Seit einigen Jahren weiß man, dass der insulinabhängige GLUT-4 auch für die Durchlässigkeit der Blut-Hirn-Schranke und die Versorgung von Gehirnzellen mit Glukose verantwortlich ist. Dies betrifft insbesondere die Nervenzellen des Hippocampus, in denen Gedächtnis- und Lernprozesse mit hoher Geschwindigkeit ablaufen [31]. Deshalb gilt Insulinresistenz heute als zentraler Faktor für die Entstehung der Alzheimer-Demenz, die als Typ-3-Diabetes bezeichnet wird [32]. Der Glukosemangel lässt Nervenzellen absterben und das Gehirn schrumpfen.
Aus diesem Grund profitieren Alzheimer-Patienten von der Umstellung auf eine ketogene Ernährung – zumindest am Anfang: In Studien verbesserte sich die Gedächtnisleistung ketogen ernährter Alzheimer-Patienten wenige Wochen nach der Ernährungsumstellung [33,34]. Die Studiendauern sind jedoch zu kurz und die Teilnehmerzahlen zu gering, um die langfristigen Auswirkungen auf das Fortschreiten der Krankheit beurteilen zu können. Erschwerend kommt hinzu, dass viele Probanden die Diät nicht lange durchhalten [35].
Macht Zucker dick?
NEIN! Zucker – auch der viel gescholtene weiße Haushaltszucker – macht einen gesunden Menschen nicht dick oder gar krank. Im Gegenteil – Glukose verbessert die mitochondriale Atmung und sorgt für die maximale ATP-Ausbeute. Dieser energetische Vorteil kommt nicht nur beim Sport, sondern auch beim Aufbau und der Regeneration von Körpergewebe zum Tragen. Neben Proteinen spielen Kohlenhydrate eine wichtige Rolle für sämtliche anabole Prozesse wie den Muskelaufbau, das Haarwachstum oder die Wundheilung.
Der anfängliche Gewichtsverlust unter Low/No Carb liegt hauptsächlich am Wasserverlust. Anders als Kohlenhydrate (Glykogen bindet das Dreifache seines Gewichts an Wasser) binden Fette kein Wasser. Langfristig reduzieren Ketone den Appetit, so dass die meisten Menschen weniger Kalorien konsumieren und abnehmen. Der Preis für die Gewichtsabnahme ist ein langsamer Stoffwechsel. Man kann den Effekt an Menschen beobachten, die mehrfach mit radikalen Diäten Gewicht verloren haben. Meistens werden diese Menschen nach jeder Diät dicker oder geraten in eine Spirale aus immer weniger Essen und immer exzessiverem Sport, um überhaupt noch abzunehmen.
Heißt das, dass wir Kohlenhydrate essen und dabei abnehmen können? JA! Mit kleinen Einschränkungen: Anders als Honig oder Obst enthält Haushaltszucker keine Nährstoffe. Wer viel Zucker konsumiert muss also auf eine gute Vitamin- und Mineralstoffversorgung achten, weil die gesteigerte mitochondrielle Atmung viele Cofaktoren verbraucht. Aber auch Vollkorngetreide oder Bohnen können Probleme machen: Unvollständig verdaut können sie eine bakterielle Fehlbesiedlung oder Darmentzündung verursachen.
Meine Erfahrungen
Mehrere Jahre lang war ich überzeugte Anhängerin von Low Carb und ketogener Ernährung. Ich konnte mich satt essen und gleichzeitig abnehmen. Am Anfang lief es auch beim Kraftsport noch relativ gut, vielleicht weil ich damals viele Kohlenhydrate in Form von Obst und Gemüse zu mir nahm.
Mit der Zeit musste ich mich mehr und mehr einschränken, weil ich wegen meiner Histaminintoleranz immer weniger Lebensmittel vertrug. Egal ob Nüsse, Tomaten, Kakao, Himbeeren, Avocado oder Schweinefleisch – nach und nach fiel immer mehr von meinem Speiseplan. Irgendwann lebte ich gefühlt nur noch von gebratenem Rindfleisch, Kokosfett und Sahne. Ich hatte kaum noch Appetit und würgte das ewig gleiche Essen mit wachsendem Widerwillen runter.
Schlimmer als der Gedanke an meine nächste Mahlzeit war nur noch die Kälte. Ich fror fast die ganze Zeit. Wenn ich mich bewegte, schwitzte ich umso mehr, nur um anschließend noch stärker zu frieren. Beim Sport hangelte ich mich von einem Leistungseinbruch zum nächsten. Es kam wie es kommen musste. Ich gab auf und aß wieder Kohlenhydrate. Ab da ging es wieder bergauf… mit meinem Körpergewicht aber vor allem mit meinem Wohlbefinden.
Rückblickend kann ich sagen, dass ich meiner Schilddrüse mit Low Carb den Rest gegeben habe. Es hat Jahre gedauert, bis meine innere Wärme zurückgekehrt ist. Auslöser für meine Schilddrüsenprobleme war zwar nicht Low Carb, sondern massiver Stress aber Low Carb hat meine Symptome definitiv verschlechtert. Für mich kommt eine kohlenhydratarme Ernährung deshalb nicht mehr infrage.
* Als Amazon-Partner verdiene ich an qualifizierten Verkäufen
Quellen
- Georg Löffler. Basiswissen Biochemie: mit Pathobiochemie. Herausgeber: Springer Buch*
- Kelley DE, Goodpaster BH, Storlien L. Muscle triglyceride and insulin resistance. Annu Rev Nutr. 2002;22:325-46
- https://www.medicoconsult.de/rt3-reverses-trijodthyronin/
- RANDLE PJ, GARLAND PB, HALES CN, NEWSHOLME EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963 Apr 13;1(7285):785-9
- Ukropcova B, Sereda O, de Jonge L, Bogacka I, Nguyen T, Xie H, Bray GA, Smith SR. Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes. 2007 Mar;56(3):720-7
- Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009 Sep;297(3):E578-91
- Turcotte LP, Fisher JS. Skeletal muscle insulin resistance: roles of fatty acid metabolism and exercise. Phys Ther. 2008 Nov;88(11):1279-96.
- Armoni M, Harel C, Bar-Yoseph F, Milo S, Karnieli E. Free fatty acids repress the GLUT4 gene expression in cardiac muscle via novel response elements. J Biol Chem. 2005 Oct 14;280(41):34786-95
- Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998 Dec;14(4):263-83
- Sugden, M. (2007). In appreciation of Sir Philip Randle: The glucose-fatty acid cycle. British Journal of Nutrition, 97(5), 809-813
- Liu M, Liu C, Zhang Z, Zhou C, Li Q, He P, Zhang Y, Li H, Qin X. Quantity and variety of food groups consumption and the risk of diabetes in adults: A prospective cohort study. Clin Nutr. 2021 Dec;40(12):5710-5717
- Lee HA, Park H. Substitution of Carbohydrates for Fats and Risk of Type 2 Diabetes among Korean Middle-Aged Adults: Findings from the Korean Genome and Epidemiology Study. Nutrients. 2022 Feb 3;14(3):654
- DocCheck Flexikon – Respiratorischer Quotient
- Westerterp KR. Food quotient, respiratory quotient, and energy balance. Am J Clin Nutr. 1993 May;57(5 Suppl):759S-764S; discussion 764S-765S
- Clarke DD, Sokoloff L. Regulation of Cerebral Metabolic Rate. In: Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999
- https://flexikon.doccheck.com/de/Zerebraler_Gefäßwiderstand
- Veselá A, Wilhelm J. The role of carbon dioxide in free radical reactions of the organism. Physiol Res. 2002;51(4):335-9
- Huskisson E, Maggini S, Ruf M. The role of vitamins and minerals in energy metabolism and well-being. J Int Med Res. 2007 May-Jun;35(3):277-89.
- Soppert J, Lehrke M, Marx N, Jankowski J, Noels H. Lipoproteins and lipids in cardiovascular disease: from mechanistic insights to therapeutic targeting. Adv Drug Deliv Rev. 2020;159:4-33
- Schönfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013 Oct;33(10):1493-9
- Turton N, Cufflin N, Dewsbury M, Fitzpatrick O, Islam R, Watler LL, McPartland C, Whitelaw S, Connor C, Morris C, Fang J, Gartland O, Holt L, Hargreaves IP. The Biochemical Assessment of Mitochondrial Respiratory Chain Disorders. Int J Mol Sci. 2022 Jul 5;23(13):7487
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009 Jan 1;417(1):1-13.
- Bisschop PH, Sauerwein HP, Endert E, Romijn JA. Isocaloric carbohydrate deprivation induces protein catabolism despite a low T3-syndrome in healthy men. Clin Endocrinol (Oxf). 2001 Jan;54(1):75-80
- Weiss EP, Villareal DT, Racette SB, Steger-May K, Premachandra BN, Klein S, Fontana L. Caloric restriction but not exercise-induced reductions in fat mass decrease plasma triiodothyronine concentrations: a randomized controlled trial. Rejuvenation Res. 2008 Jun;11(3):605-9
- Iacovides S, Maloney SK, Bhana S, Angamia Z, Meiring RM. Could the ketogenic diet induce a shift in thyroid function and support a metabolic advantage in healthy participants? A pilot randomized-controlled-crossover trial. PLoS One. 2022 Jun 3;17(6):e0269440
- Wikipedia – Ketogene Diät
- Nakao R, Abe T, Yamamoto S, Oishi K. Ketogenic diet induces skeletal muscle atrophy via reducing muscle protein synthesis and possibly activating proteolysis in mice. Sci Rep. 2019 Dec 23;9(1):19652
- Baskin KK, Taegtmeyer H. Taking pressure off the heart: the ins and outs of atrophic remodelling. Cardiovasc Res. 2011 May 1;90(2):243-50
- Yakupova EI, Bocharnikov AD, Plotnikov EY. Effects of Ketogenic Diet on Muscle Metabolism in Health and Disease. Nutrients. 2022 Sep 16;14(18):3842
- Melkonian EA, Asuka E, Schury MP. Physiology, Gluconeogenesis. [Updated 2022 May 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan
- McNay EC, Pearson-Leary J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp Neurol. 2020 Jan;323:113076
- Kandimalla R, Thirumala V, Reddy PH. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim Biophys Acta Mol Basis Dis. 2017 May;1863(5):1078-1089
- Rusek M, Pluta R, Ułamek-Kozioł M, Czuczwar SJ. Ketogenic Diet in Alzheimer’s Disease. Int J Mol Sci. 2019 Aug 9;20(16):3892
- Phillips MCL, Deprez LM, Mortimer GMN, Murtagh DKJ, McCoy S, Mylchreest R, Gilbertson LJ, Clark KM, Simpson PV, McManus EJ, Oh JE, Yadavaraj S, King VM, Pillai A, Romero-Ferrando B, Brinkhuis M, Copeland BM, Samad S, Liao S, Schepel JAC. Randomized crossover trial of a modified ketogenic diet in Alzheimer’s disease. Alzheimers Res Ther. 2021 Feb 23;13(1):51
- https://www.businessinsider.com/why-keto-diet-is-hard-2019-5




{kind=link}
Comments powered by Talkyard.